 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSingle cell multi omics data
Papers and Code
Open World Knowledge Aided Single-Cell Foundation Model with Robust Cross-Modal Cell-Language Pre-training
Jan 09, 2026Recent advancements in single-cell multi-omics, particularly RNA-seq, have provided profound insights into cellular heterogeneity and gene regulation. While pre-trained language model (PLM) paradigm based single-cell foundation models have shown promise, they remain constrained by insufficient integration of in-depth individual profiles and neglecting the influence of noise within multi-modal data. To address both issues, we propose an Open-world Language Knowledge-Aided Robust Single-Cell Foundation Model (OKR-CELL). It is built based on a cross-modal Cell-Language pre-training framework, which comprises two key innovations: (1) leveraging Large Language Models (LLMs) based workflow with retrieval-augmented generation (RAG) enriches cell textual descriptions using open-world knowledge; (2) devising a Cross-modal Robust Alignment (CRA) objective that incorporates sample reliability assessment, curriculum learning, and coupled momentum contrastive learning to strengthen the model's resistance to noisy data. After pretraining on 32M cell-text pairs, OKR-CELL obtains cutting-edge results across 6 evaluation tasks. Beyond standard benchmarks such as cell clustering, cell-type annotation, batch-effect correction, and few-shot annotation, the model also demonstrates superior performance in broader multi-modal applications, including zero-shot cell-type annotation and bidirectional cell-text retrieval.
You Only Train Once: Differentiable Subset Selection for Omics Data
Dec 19, 2025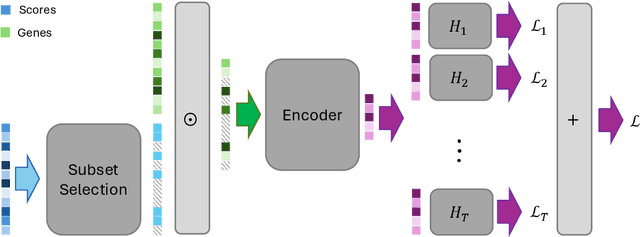

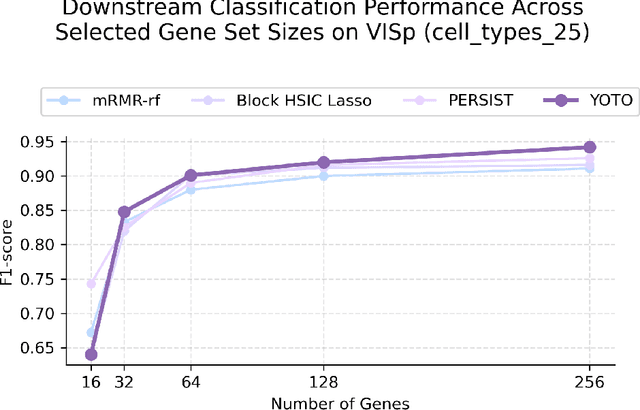
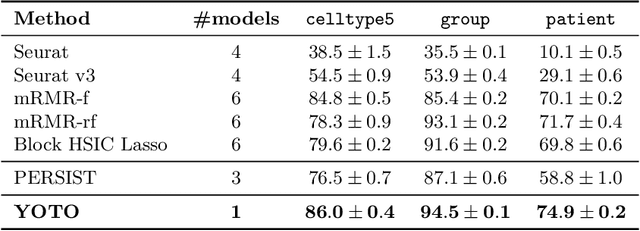
Selecting compact and informative gene subsets from single-cell transcriptomic data is essential for biomarker discovery, improving interpretability, and cost-effective profiling. However, most existing feature selection approaches either operate as multi-stage pipelines or rely on post hoc feature attribution, making selection and prediction weakly coupled. In this work, we present YOTO (you only train once), an end-to-end framework that jointly identifies discrete gene subsets and performs prediction within a single differentiable architecture. In our model, the prediction task directly guides which genes are selected, while the learned subsets, in turn, shape the predictive representation. This closed feedback loop enables the model to iteratively refine both what it selects and how it predicts during training. Unlike existing approaches, YOTO enforces sparsity so that only the selected genes contribute to inference, eliminating the need to train additional downstream classifiers. Through a multi-task learning design, the model learns shared representations across related objectives, allowing partially labeled datasets to inform one another, and discovering gene subsets that generalize across tasks without additional training steps. We evaluate YOTO on two representative single-cell RNA-seq datasets, showing that it consistently outperforms state-of-the-art baselines. These results demonstrate that sparse, end-to-end, multi-task gene subset selection improves predictive performance and yields compact and meaningful gene subsets, advancing biomarker discovery and single-cell analysis.
Modeling Dabrafenib Response Using Multi-Omics Modality Fusion and Protein Network Embeddings Based on Graph Convolutional Networks
Dec 13, 2025Cancer cell response to targeted therapy arises from complex molecular interactions, making single omics insufficient for accurate prediction. This study develops a model to predict Dabrafenib sensitivity by integrating multiple omics layers (genomics, transcriptomics, proteomics, epigenomics, and metabolomics) with protein network embeddings generated using Graph Convolutional Networks (GCN). Each modality is encoded into low dimensional representations through neural network preprocessing. Protein interaction information from STRING is incorporated using GCN to capture biological topology. An attention based fusion mechanism assigns adaptive weights to each modality according to its relevance. Using GDSC cancer cell line data, the model shows that selective integration of two modalities, especially proteomics and transcriptomics, achieves the best test performance (R2 around 0.96), outperforming all single omics and full multimodal settings. Genomic and epigenomic data were less informative, while proteomic and transcriptomic layers provided stronger phenotypic signals related to MAPK inhibitor activity. These results show that attention guided multi omics fusion combined with GCN improves drug response prediction and reveals complementary molecular determinants of Dabrafenib sensitivity. The approach offers a promising computational framework for precision oncology and predictive modeling of targeted therapies.
MoRE-GNN: Multi-omics Data Integration with a Heterogeneous Graph Autoencoder
Oct 08, 2025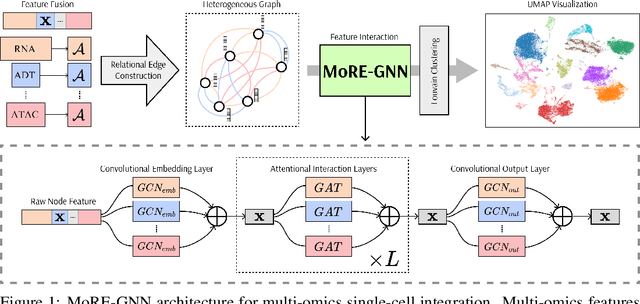

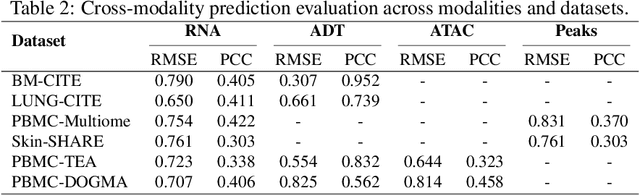
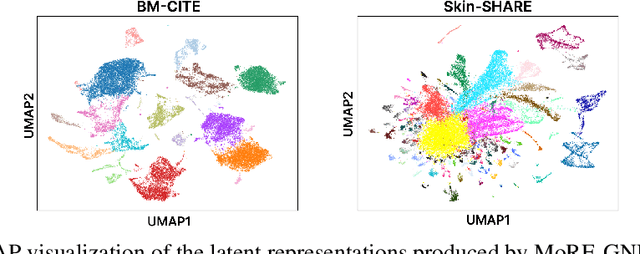
The integration of multi-omics single-cell data remains challenging due to high-dimensionality and complex inter-modality relationships. To address this, we introduce MoRE-GNN (Multi-omics Relational Edge Graph Neural Network), a heterogeneous graph autoencoder that combines graph convolution and attention mechanisms to dynamically construct relational graphs directly from data. Evaluations on six publicly available datasets demonstrate that MoRE-GNN captures biologically meaningful relationships and outperforms existing methods, particularly in settings with strong inter-modality correlations. Furthermore, the learned representations allow for accurate downstream cross-modal predictions. While performance may vary with dataset complexity, MoRE-GNN offers an adaptive, scalable and interpretable framework for advancing multi-omics integration.
LLM4Cell: A Survey of Large Language and Agentic Models for Single-Cell Biology
Oct 09, 2025Large language models (LLMs) and emerging agentic frameworks are beginning to transform single-cell biology by enabling natural-language reasoning, generative annotation, and multimodal data integration. However, progress remains fragmented across data modalities, architectures, and evaluation standards. LLM4Cell presents the first unified survey of 58 foundation and agentic models developed for single-cell research, spanning RNA, ATAC, multi-omic, and spatial modalities. We categorize these methods into five families-foundation, text-bridge, spatial, multimodal, epigenomic, and agentic-and map them to eight key analytical tasks including annotation, trajectory and perturbation modeling, and drug-response prediction. Drawing on over 40 public datasets, we analyze benchmark suitability, data diversity, and ethical or scalability constraints, and evaluate models across 10 domain dimensions covering biological grounding, multi-omics alignment, fairness, privacy, and explainability. By linking datasets, models, and evaluation domains, LLM4Cell provides the first integrated view of language-driven single-cell intelligence and outlines open challenges in interpretability, standardization, and trustworthy model development.
CellVerse: Do Large Language Models Really Understand Cell Biology?
May 09, 2025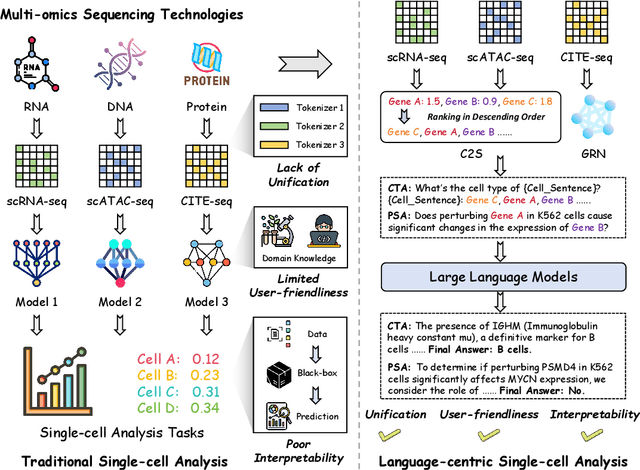
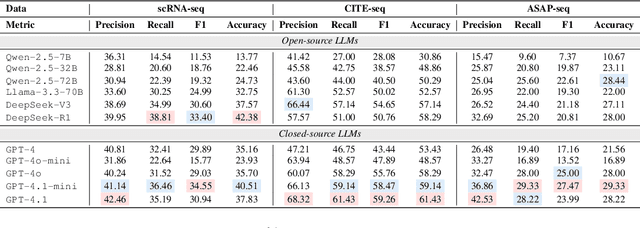
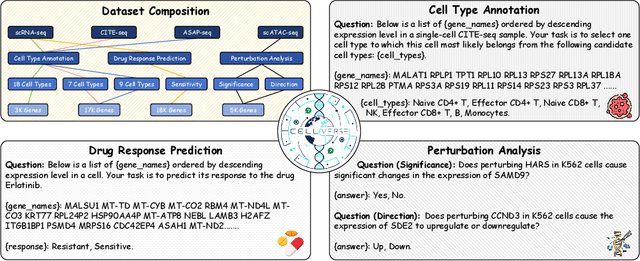
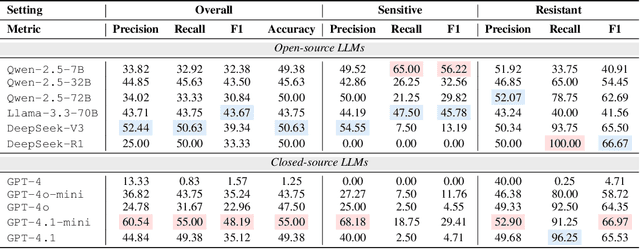
Recent studies have demonstrated the feasibility of modeling single-cell data as natural languages and the potential of leveraging powerful large language models (LLMs) for understanding cell biology. However, a comprehensive evaluation of LLMs' performance on language-driven single-cell analysis tasks still remains unexplored. Motivated by this challenge, we introduce CellVerse, a unified language-centric question-answering benchmark that integrates four types of single-cell multi-omics data and encompasses three hierarchical levels of single-cell analysis tasks: cell type annotation (cell-level), drug response prediction (drug-level), and perturbation analysis (gene-level). Going beyond this, we systematically evaluate the performance across 14 open-source and closed-source LLMs ranging from 160M to 671B on CellVerse. Remarkably, the experimental results reveal: (1) Existing specialist models (C2S-Pythia) fail to make reasonable decisions across all sub-tasks within CellVerse, while generalist models such as Qwen, Llama, GPT, and DeepSeek family models exhibit preliminary understanding capabilities within the realm of cell biology. (2) The performance of current LLMs falls short of expectations and has substantial room for improvement. Notably, in the widely studied drug response prediction task, none of the evaluated LLMs demonstrate significant performance improvement over random guessing. CellVerse offers the first large-scale empirical demonstration that significant challenges still remain in applying LLMs to cell biology. By introducing CellVerse, we lay the foundation for advancing cell biology through natural languages and hope this paradigm could facilitate next-generation single-cell analysis.
ChromFound: Towards A Universal Foundation Model for Single-Cell Chromatin Accessibility Data
May 19, 2025The advent of single-cell Assay for Transposase-Accessible Chromatin using sequencing (scATAC-seq) offers an innovative perspective for deciphering regulatory mechanisms by assembling a vast repository of single-cell chromatin accessibility data. While foundation models have achieved significant success in single-cell transcriptomics, there is currently no foundation model for scATAC-seq that supports zero-shot high-quality cell identification and comprehensive multi-omics analysis simultaneously. Key challenges lie in the high dimensionality and sparsity of scATAC-seq data, as well as the lack of a standardized schema for representing open chromatin regions (OCRs). Here, we present \textbf{ChromFound}, a foundation model tailored for scATAC-seq. ChromFound utilizes a hybrid architecture and genome-aware tokenization to effectively capture genome-wide long contexts and regulatory signals from dynamic chromatin landscapes. Pretrained on 1.97 million cells from 30 tissues and 6 disease conditions, ChromFound demonstrates broad applicability across 6 diverse tasks. Notably, it achieves robust zero-shot performance in generating universal cell representations and exhibits excellent transferability in cell type annotation and cross-omics prediction. By uncovering enhancer-gene links undetected by existing computational methods, ChromFound offers a promising framework for understanding disease risk variants in the noncoding genome.
Subset-Contrastive Multi-Omics Network Embedding
Apr 15, 2025



Motivation: Network-based analyses of omics data are widely used, and while many of these methods have been adapted to single-cell scenarios, they often remain memory- and space-intensive. As a result, they are better suited to batch data or smaller datasets. Furthermore, the application of network-based methods in multi-omics often relies on similarity-based networks, which lack structurally-discrete topologies. This limitation may reduce the effectiveness of graph-based methods that were initially designed for topologies with better defined structures. Results: We propose Subset-Contrastive multi-Omics Network Embedding (SCONE), a method that employs contrastive learning techniques on large datasets through a scalable subgraph contrastive approach. By exploiting the pairwise similarity basis of many network-based omics methods, we transformed this characteristic into a strength, developing an approach that aims to achieve scalable and effective analysis. Our method demonstrates synergistic omics integration for cell type clustering in single-cell data. Additionally, we evaluate its performance in a bulk multi-omics integration scenario, where SCONE performs comparable to the state-of-the-art despite utilising limited views of the original data. We anticipate that our findings will motivate further research into the use of subset contrastive methods for omics data.
Learning to Match Unpaired Data with Minimum Entropy Coupling
Mar 11, 2025Multimodal data is a precious asset enabling a variety of downstream tasks in machine learning. However, real-world data collected across different modalities is often not paired, which is a significant challenge to learn a joint distribution. A prominent approach to address the modality coupling problem is Minimum Entropy Coupling (MEC), which seeks to minimize the joint Entropy, while satisfying constraints on the marginals. Existing approaches to the MEC problem focus on finite, discrete distributions, limiting their application for cases involving continuous data. In this work, we propose a novel method to solve the continuous MEC problem, using well-known generative diffusion models that learn to approximate and minimize the joint Entropy through a cooperative scheme, while satisfying a relaxed version of the marginal constraints. We empirically demonstrate that our method, DDMEC, is general and can be easily used to address challenging tasks, including unsupervised single-cell multi-omics data alignment and unpaired image translation, outperforming specialized methods.
Benchmarking AI scientists in omics data-driven biological research
May 13, 2025The rise of large language models and multi-agent systems has sparked growing interest in AI scientists capable of autonomous biological research. However, existing benchmarks either focus on reasoning without data or on data analysis with predefined statistical answers, lacking realistic, data-driven evaluation settings. Here, we introduce the Biological AI Scientist Benchmark (BaisBench), a benchmark designed to assess AI scientists' ability to generate biological discoveries through data analysis and reasoning with external knowledge. BaisBench comprises two tasks: cell type annotation on 31 expert-labeled single-cell datasets, and scientific discovery through answering 198 multiple-choice questions derived from the biological insights of 41 recent single-cell studies. Systematic experiments on state-of-the-art AI scientists and LLM agents showed that while promising, current models still substantially underperform human experts on both tasks. We hope BaisBench will fill this gap and serve as a foundation for advancing and evaluating AI models for scientific discovery. The benchmark can be found at: https://github.com/EperLuo/BaisBench.